Why ultrafast laser spectroscopy?
Fast Reaction Kinetics: The simplest way to determine the rate of a chemical reaction is to mix the reagents and follow changes as a function of time using UV/vis absorption or some other spectroscopy. But what if the reaction is complete as soon you mix the reagents? In such cases, a faster technique is necessitated, such as the stopped-flow method, which enables data collection just milliseconds after mixing the starting materials using syringe-controlled reservoirs. For reactions that occur too quickly for even the stopped-flow method, time-resolved laser spectroscopy is utilized. For example, our group uses ultrafast pump-probe (transient absorption) laser spectroscopy to study some of the fastest phenomena that occur in chemistry, with instrumentation that enables a time resolution of 200 femtoseconds (1 fs = 10-15 s).
A beautiful example of such ultrafast studies is the photochemistry of rhodopsin, a protein involved in our vision. As you read this sentence, numerous retinal molecules (prosthetic group of rhodopsin) in your eyes undergo an ultrafast, sub-picosecond cis-trans photoisomerization, a process that triggers a cascade of biochemical responses that ultimately lead to your visual perception. The dynamics of retinal photoisomerization was studied in 2010 by Cerullo et al. using ultrafast spectroscopy to temporally map the cis-to-trans transition (Figure 1). The transient absorption spectra show a temporal evolution from blue signal (due to stimulated emission from the excited cis-retinal) to the red signal (due to the ground-state absorption of the trans-retinal). The reaction occurs within 200 fs!
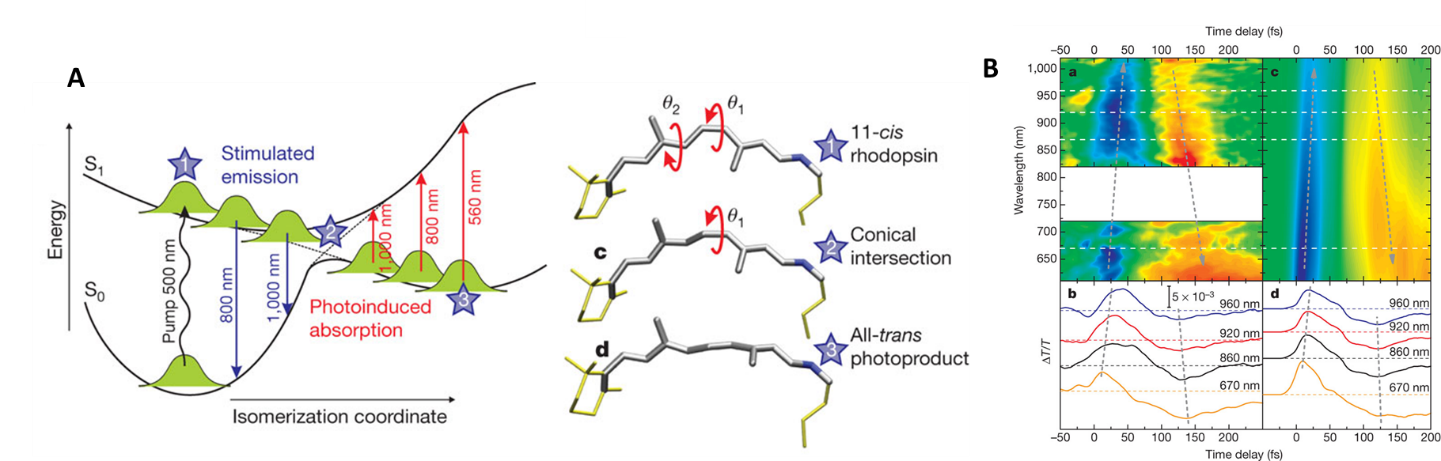
Figure 1: Rhodopsin photoisomerization studied using ultrafast laser spectroscopy (Cerullo, Nature 2010): (A) Potential energy surfaces and structures; (B) experimental and simulated transient absorption spectra of rhodopsin.
Structural Information: The previous example illustrates how transients formed after the photo-excitation pulse can be observed using UV/Vis absorption spectroscopy. Unfortunately, electronic absorption does not provide significant structural information about the short-lived intermediates. An alternative approach is to use mid-IR laser pulse to probe the transients. The absorption bands in the IR region are skinny and less likely to overlap, enabling the structural characterization of multiple species or states. Furthermore, the band maxima (vibrational frequencies) provides information about the bond types and lengths in the transient.
For example, our group investigated the structural changes that take place when a Re-based complex is photoexcited at 500 nm to form the metal-to-ligand charge transfer (MLCT) excited state (Figure 2). The carbonyl group vibrational frequencies shift to higher wavenumbers upon-photoexcitation, showing that CO bonds become stronger in the MLCT state. The aromatic ligand frequencies show changes only in the tetrazine ring vibrations, while the pyridine vibrations do not change upon excitation. This result is consistent with electron being delocalized only across the tetrazine moiety and matches the DFT calculations.
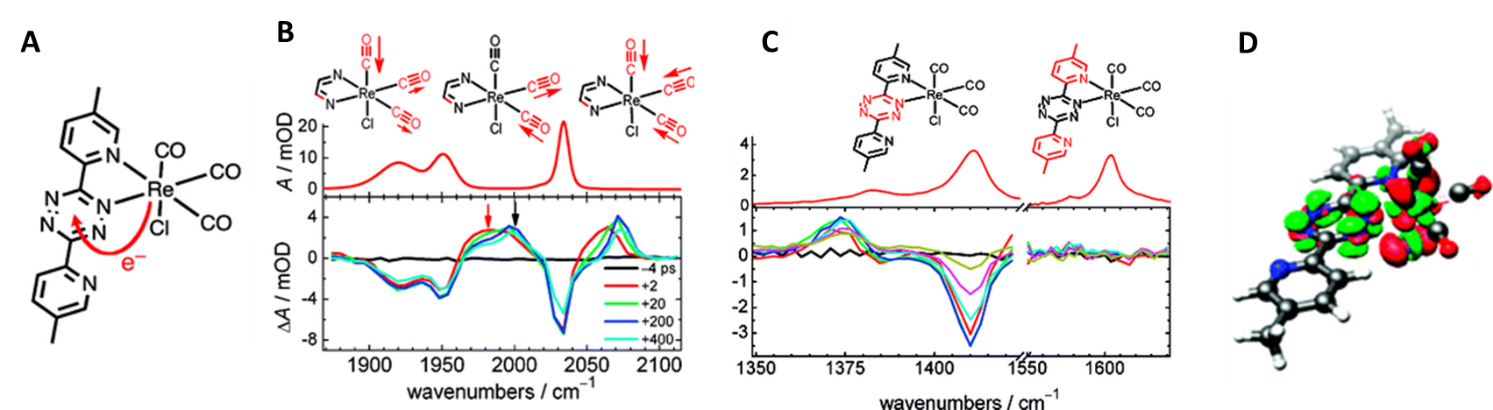
Figure 2: Structural changes associated with the formation of MLCT state in Re-based complex (Glusac, JACS 2009). (A) Structure of Re(CO)3Cl(Me2BPTZ) and the electron transfer direction that takes place when MLCT excited state is formed; (B) Steady-state and time-resolved IR spectroscopy of the carbonyl-region. The vibrational frequencies shift to higher wavenumbers upon excitation, consistent with the CO bond strengthening due to the oxidation of the Re-center; (C) IR spectra in the ligand region. The time-resolved spectrum shows changes only in the tetrazine ring vibrations, indicating that the electron does not delocalized along the pyridine moieties; (D) Calculated difference density plot at the B3LYP level of theory (red/green = depletion/accumulation of charge).
Ultrafast Microscopy: Another interesting application of ultrafast lasers is transient absorption microscopy (TAM). Similar to ultrafast spectroscopy, TAM is a time-resolved method that illustrates the evolution of a system in short time intervals (pico- and femtosecond time domains) by using pulsed pump and probe lasers. In addition to conventional spectroscopy, TAM also enables spatially resolved experiments with resolution of a few hundred nanometers. For example, TAM was used to study ligth harvesting in tubular porphyrin aggregates (Figure 3) by the Huang group and the study enabled the direct observation of time-resolved exciton propagation and transport along the chromophore assembly.
Figure 3a shows the direct imaging of a transient signal probed at the ground-state bleach wavelength at different time delays (0, 3 and 15 ps). The observed bleach signal becomes broader at longer time delays, which is consistent with the excitons migrating along the material, away from the location where they were initially formed at time zero by the pump beam. Figure 3b is a cross-sectional representation of exciton diffusion as a function of probe delay. The experimental data was used to derive the exciton diffusion constant (D=3-6 cm2/s) in porphyrin chromophore assemblies.
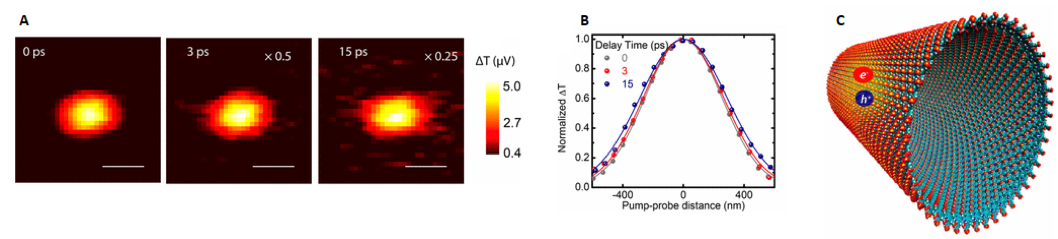
Figure 3: Direct imaging of exciton formation and diffusion via transient absorption microscopy (Huang, JACS, 2017). (A) Experimentally obtained images of exciton diffusion at different pump-probe time delays. (B) Cross-sections of the TAM images at each time delay. (C) Computer generated figure of a tubular porphyrin aggregate with exciton propagation occurring on the outer surface.